Kreepa G Kooblall gave the 2023 Early Career Prize Lecture (Science) at SfE BES 2023 in Glasgow. We are delighted to include this summary of her award-winning work here.
Skeletal dysplasia syndromes are rare genetic disorders caused by the abnormal development of bone and cartilage. Currently, more than 461 heterogeneous skeletal dysplasia syndromes have been reported. While individually they are rare, together they account for an estimated overall prevalence of ~2.3 cases per 10,000 live births.1,2
'In addition to helping identify new features in patients with MSS that should be monitored as part of their routine care, the NfixDel2/Del2 mice have also been useful in identifying potential therapeutic targets for MSS.'
Skeletal dysplasia syndromes normally first present at a young age. Their prenatal diagnosis is often difficult, due to heterogeneity both within and among disorders. Moreover, because of their rarity, clinical investigations into these disorders remain challenging, as do finding possible treatments and cures.1,2
MARSHALL–SMITH SYNDROME, MALAN SYNDROME AND NFIX VARIANTS
Marshall–Smith syndrome (MSS) and Malan syndrome (MAL) are two examples of congenital skeletal dysplasia syndromes. The skeletal and neural abnormalities in patients with MSS or MAL are caused by heterozygous mutations affecting the nuclear factor I/X (NFIX) gene.3,4
MSS is caused by frameshift mutations that affect the C-terminal part of the NFIX gene, leading to aberrant transcripts escaping the nonsense decay mechanism (NMD) and resulting in the production of dysfunctional mutant NFIX proteins that are believed to behave in a dominant negative manner.3,4 In contrast, MAL is due to mutations that predominantly affect the N-terminal part of the NFIX gene, leading to NFIX haploinsufficiency.3,4 NFIX is a transcription factor which plays an important role in the regulation of gene expression during the development of many organs and tissues (including the skeleton and nervous system).5
THE IMPORTANCE OF MOUSE MODELS FOR RARE GENETIC DISORDERS
Since NFIX is a ubiquitously expressed protein, and because MSS and MAL are rare multisystem disorders, it is difficult to obtain tissue samples from the patients, who are mainly children with mental and physical impairments living in different countries. Therefore, to circumvent this problem and to find a common solution for MSS and MAL, we have generated a mouse model for MSS (since it is the more severe of the two disorders). This enables us to study the effects of mutant NFIX and understand the mechanisms underlying the aetiology of MSS, as well as providing a useful tool for investigating possible treatments for MSS, and potentially MAL.
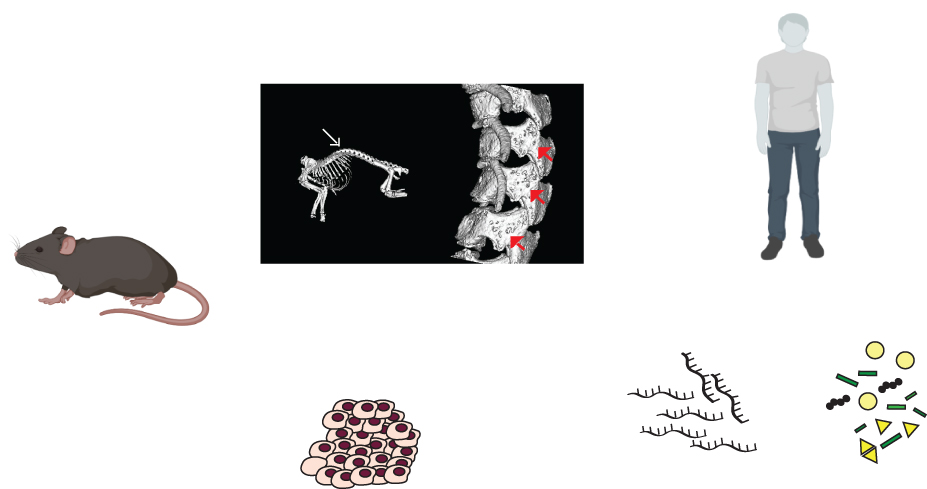
In order to generate a mouse model for MSS, we used the CRISPR–Cas9 gene editing system to target a two-nucleotide deletion in Nfix exon 7, which is the most commonly mutated exon in patients with MSS. We used in vitro expression assays to demonstrate that this frameshift mutation was not cleared by the NMD and led to the production of a dysfunctional mutant NFIX protein with aberrant function.6 We therefore characterised this Nfix Del2 mouse model for features of MSS and showed that, in contrast to patients with MSS who are heterozygous for NFIX mutations, the heterozygous Nfix+/Del2 mouse was viable, normal and fertile. In contrast, the homozygous NfixDel2/Del2 mouse had short stature and developmental delays, as well as skeletal (including kyphosis, osteopenia, reduced bone mineral content), cranial, neural (due to abnormal brain morphologies), hepatic and renal abnormalities.6 However, phenotypic differences between organisms are not uncommon, and can be attributed to allelic variation, modifier genes, genetic variations, genetic background and environmental conditions in animal models versus in patients.7
Thus, NfixDel2/Del2 mice may represent a mouse model for MSS, in which patients commonly have short stature, developmental delays, skeletal abnormalities (including craniofacial defects, osteopenia with increased fracture rate, kyphoscoliosis, decreased bone density8) and intellectual disability due to non-specific brain abnormalities.3,9,10 Moreover, following our identification of likely renal dysfunction in the NfixDel2/Del2 mice, investigations were undertaken by ultrasound in two patients with MSS, and this revealed the occurrence of renal cysts and nephrocalcinosis in these cases.6

Figure. A mouse model for MSS has helped identify new features in patients which should be monitored as part of their care, as well as new genes and pathways which are altered, that could potentially be therapeutic targets for MSS and MAL. qRT−PCR, quantitative reverse transcription−polymerase chain reaction.
IDENTIFYING POTENTIAL THERAPEUTIC TARGETS FOR MSS AND MAL
In addition to helping identify new features in patients with MSS that should be monitored as part of their routine care, the NfixDel2/Del2 mice have also been useful in identifying potential therapeutic targets for MSS.
We used mouse embryonic fibroblast cells (MEFs) derived from the Nfix Del2 mice for RNA sequencing and proteomic analyses, in order to identify genes and pathways that are misregulated in patients with MSS. We chose to initially undertake RNA sequencing and proteomics studies in MEFs, because mice generated from the same genetic background show reduced genotypic variability compared with fibroblasts derived from unrelated patients with MSS. This approach therefore maximised our chances of identifying statistically significantly altered genes and pathways.
Validation studies using fibroblast cells derived from patients with MSS or MAL have confirmed that two genes involved in the retinoic acid (RA) pathway were misregulated at the RNA and protein levels both in MEFs obtained from the NfixDel2/Del2 mice and in fibroblasts from patients with MSS or MAL. This may indicate a possible misregulation of the RA pathway in these patients, and the possibility that drugs targeting the RA pathway might be of therapeutic benefit to these patients with skeletal dysplasia.
KREEPA G KOOBLALL
Academic Endocrine Unit, OCDEM, University of Oxford
This work was undertaken in my role as a post-doctoral research scientist in the laboratory of Professor Rajesh Thakker (Academic Endocrine Unit, OCDEM, University of Oxford).
Find out more about Early Career Prize Lectures
REFERENCES
1. Mortier GR et al. 2019 American Journal of Medical Genetics Part A https://doi.org/10.1002/ajmg.a.61366.
2. Moy N et al. 2023 Quality of Life Research https://doi.org/10.1007/s11136-023-03431-z.
3. Malan V et al. 2010 American Journal of Human Genetics https://doi.org/10.1016%2Fj.ajhg.2010.07.001.
4. Priolo M et al. 2018 Human Mutation https://doi.org/10.1002/humu.23563.
5. Piper M et al. 2019 Trends in Cell Biology https://doi.org/10.1016/j.tcb.2018.09.003.
6. Kooblall KG et al. 2023 Jbmr Plus https://doi.org/10.1002/jbm4.10739.
7. Raj A et al. 2010 Nature https://doi.org/10.1038/nature08781.
8. Shaw AC et al. 2010 American Journal of Medical Genetics Part A https://doi.org/10.1002/ajmg.a.33709.
9. Schanze D et al. 2014 Human Mutation https://doi.org/10.1002/humu.22603.
10. Mulder PA et al. 2020 Journal of Intellectual Disability Research https://doi.org/10.1111/jir.12787.




{kind=link}